Blog
Phenylketonuria (PKU)
Covenant Metabolic Specialists Health Library
Covenant Metabolic Specialists
Physician Reviewed
Dec 3, 2025
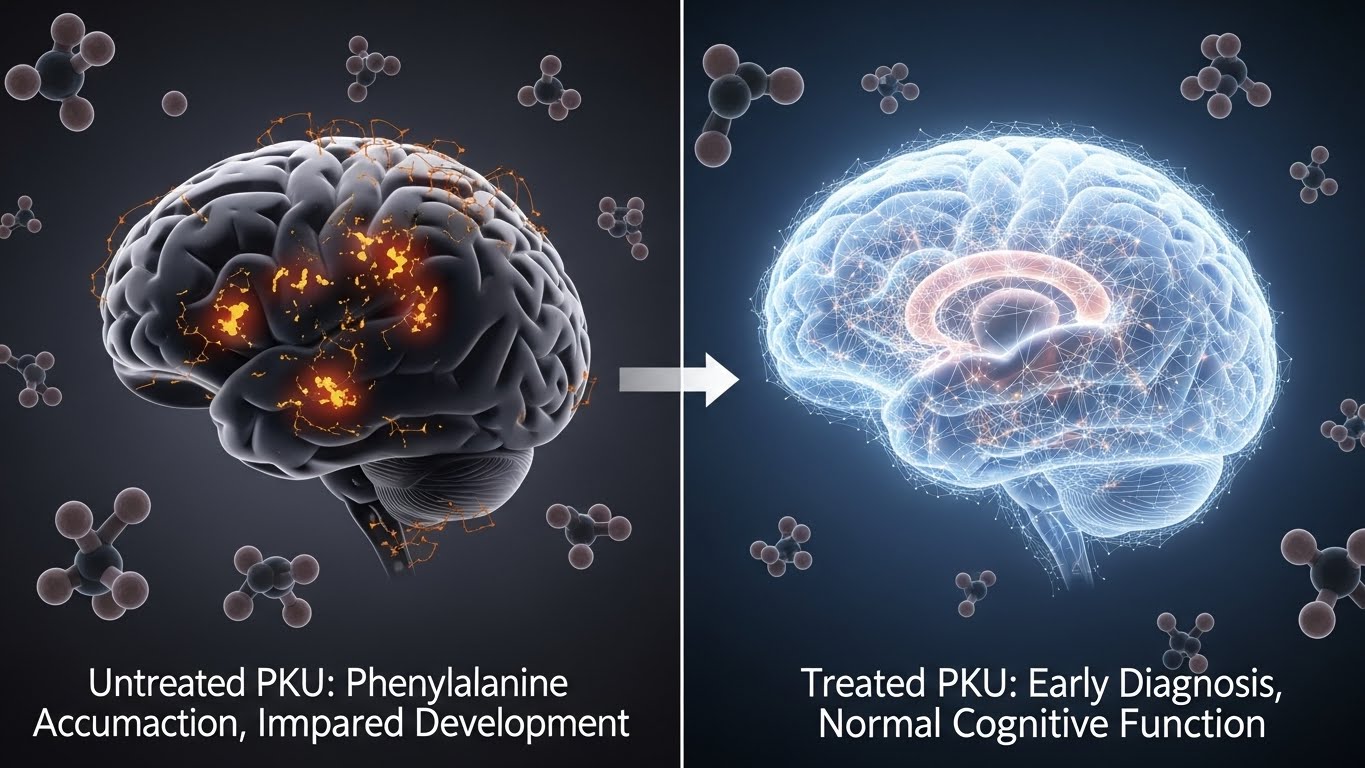
Phenylketonuria is a rare inborn error of metabolism caused by phenylalanine hydroxylase deficiency. Without enzymatic conversion, phenylalanine accumulates to neurotoxic levels, damaging developing brain tissue. Modern newborn screening identifies PKU early, allowing dietary management that prevents intellectual disability and fosters normal cognitive development and quality of life.
Symptoms
Untreated infants appear normal at birth but soon exhibit vomiting, irritability, eczema, hypopigmentation, musty body odor, and developmental delay. Seizures, microcephaly, and irreversible intellectual disability follow. When treated from infancy, patients are largely symptom‑free yet must monitor phenylalanine to avoid attention deficits and mood disorders during life transitions.
Causes
Biallelic pathogenic variants in the PAH gene cripple phenylalanine hydroxylase activity. Some cases result from defects in BH4 cofactor synthesis or recycling. High phenylalanine saturates neutral amino‑acid transporters at the blood–brain barrier, depleting tyrosine and tryptophan entry and undermining dopamine and serotonin synthesis crucial for neurodevelopment.
Risk Factors
Risk depends on having two carrier parents. Maternal PKU with poor dietary control exposes the fetus to teratogenic phenylalanine levels causing microcephaly, congenital heart disease, and growth restriction. Late diet relaxation, adolescence, and college environments risk metabolic control lapses.
Diagnosis
Universal newborn heel‑stick screening measures phenylalanine at 24–72 hours. Confirmatory quantitative amino‑acid chromatography, genotype analysis, and sapropterin response testing follow. Lifelong monitoring of phenylalanine and tyrosine guides dietary adjustments. Neuropsychological evaluations track subtle executive‑function impacts.
Treatments
Treatment centers on a lifelong low‑phenylalanine medical formula plus precisely weighed low‑protein foods. Sapropterin stimulates residual PAH in responsive genotypes, raising dietary tolerance. Pegvaliase enzyme substitution permits more liberal protein intake in adults. Regular clinic oversight ensures nutrient sufficiency and adherence.
Prevention
Strict diet from infancy prevents cognitive impairment. Pre‑pregnancy metabolic control averts fetal damage in maternal PKU. Education, mobile tracking apps, social support, and coverage for costly medical foods safeguard long‑term metabolic health.
Our Take
Covenant integrates tele‑nutrition, remote phenylalanine testing, and peer mentorship so PKU patients feel supported rather than isolated by dietary constraints.
From fatal neurodevelopmental disorder to manageable chronic condition, PKU exemplifies newborn‑screening success. Covenant ensures ongoing support as patients navigate childhood, adolescence, pregnancy, and adulthood without neurological sacrifice.
